Workflow¶
This page provides a brief explanation of the workflow to perform APR calculations with pAPRika. For more detail users are recommended to go through the tutorials, which details further on how to setup and run APR simulations from start to finish.
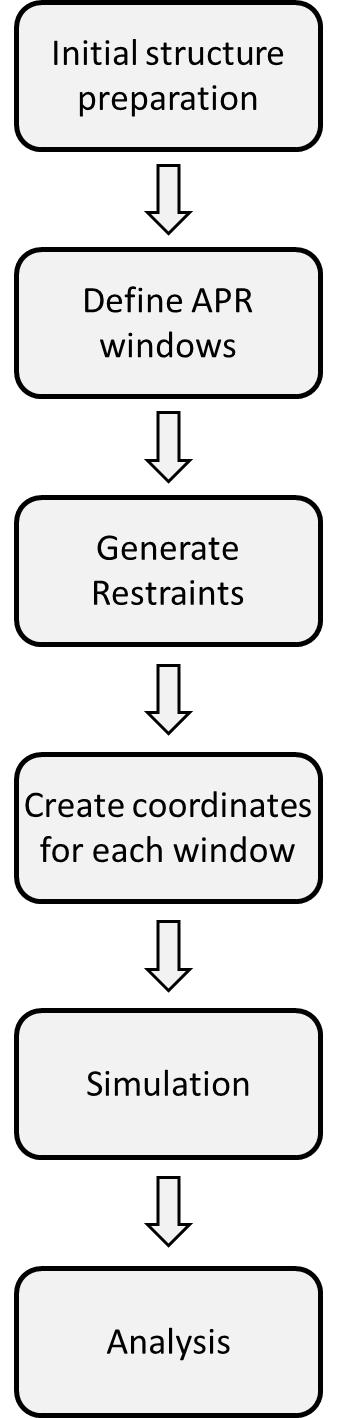
Flowchart of the paprika workflow for a typical APR simulation.¶
Structure Preparation¶
Aligning the host-guest complex
The starting structure for the APR simulation can be configured with paprika. The APR calculation is most efficient in
a rectangular box with the long axis parallel to the pulling axis (reduces the number of water molecules in the system).
To make this easy to set up, the Align module provides functions to shift and orient a structure. For example, we can
translate a structure to the origin and then orient the system to the \(z\)-axis by running
from paprika.build.align import *
translated_structure = translate_to_origin(structure)
aligned_structure = zalign(translated_structure, ":GST@C1", ":GST@C2")
Adding dummy atoms
To provide something to pull “against,” we add dummy atoms that are fixed in place with strong positional restraints.
These dummy atoms can be added to a the host-guest structure using the Dummy Atoms module in paprika.
from paprika.build.dummy import add_dummy
structure = dummy.add_dummy(structure, residue_name="DM1", z=-6.0)
structure = dummy.add_dummy(structure, residue_name="DM2", z=-9.0)
structure = dummy.add_dummy(structure, residue_name="DM3", z=-11.2, y=2.2)
structure.save("aligned_with_dummy.pdb", overwrite=True)
We will need the mol2 and frcmod files for the dummy atoms, which we will need to generate the AMBER topology
dummy.write_dummy_frcmod(filepath="complex/dummy.frcmod")
dummy.write_dummy_mol2(residue_name="DM1", filepath="complex/dm1.mol2")
dummy.write_dummy_mol2(residue_name="DM2", filepath="complex/dm2.mol2")
dummy.write_dummy_mol2(residue_name="DM3", filepath="complex/dm3.mol2")
Building the topology
Finally, we can use the tleap wrapper to combine all of these components to generate the topology and coordinate files.
from paprika.build.system import TLeap
system = TLeap()
system.output_prefix = "host-guest-dum"
system.pbc_type = None
system.neutralize = False
system.template_lines = [
"source leaprc.gaff",
"HST = loadmol2 host.mol2",
"GST = loadmol2 guest.mol2",
"DM1 = loadmol2 dm1.mol2",
"DM2 = loadmol2 dm2.mol2",
"DM3 = loadmol2 dm3.mol2",
"model = loadpdb aligned_with_dummy.pdb",
]
system.build()
Solvating the structure
The TLeap wrapper also provides an API for choosing water models when the user wants to solvate their structure.
tleap provides a number of models for both 3-point and 4-point water models. There are also sub-types for each
water model, e.g. for TIP3P we can choose to use the ForceBalance optimized variant called TIP3P-FB or the host-guest
binding optimized model called Bind3P. To choose a water model for solvation we use the set_water_model method of
the TLeap wrapper. The method requires the user to specify the water model and optionally the sub-type as the
model_type attibute. The supported water models are:
spc: None (SPCBOX), “flexible” (SPCFWBOX), “quantum” (QSPCFWBOX)
opc: None (OPCBOX), “three-point” (OPC3BOX)
tip3p: None (TIP3PBOX), “flexible” (TIP3PFBOX), “force-balance” (FB3BOX)
tip4p: None (TIP4PBOX), “ewald” (TIP4PEWBOX), “force-balance” (FB4BOX)
Below is an example for solvating a system with 2000 TIP3P water molecules with ForceBalance optimized parameters.
from paprika.build.system import TLeap
from paprika.build.system.utils import PBCBox
system = TLeap()
system.output_prefix = "host-guest-dum"
system.pbc_type = PBCBox.rectangular
system.target_waters = 2000
system.set_water_model("tip3p", model_type="force-balance")
system.template_lines = [
"source leaprc.gaff",
"HST = loadmol2 host.mol2",
"GST = loadmol2 guest.mol2",
"DM1 = loadmol2 dm1.mol2",
"DM2 = loadmol2 dm2.mol2",
"DM3 = loadmol2 dm3.mol2",
"model = loadpdb aligned_with_dummy.pdb",
]
system.build()
Defining Restraints¶
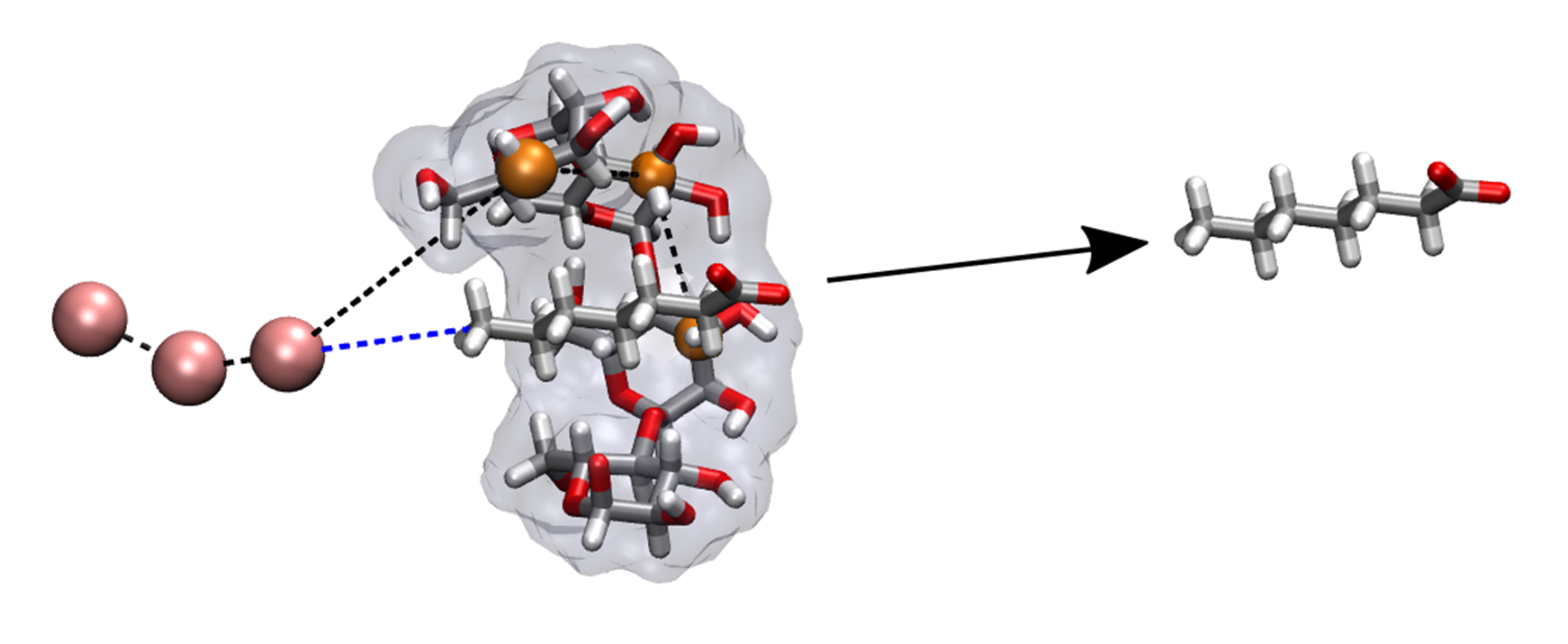
In APR calculations we apply restraints on the host (or protein) and the guest molecules. The restraints can be grouped into four categories: (1) static restraints, (2) varying restraints, (3) wall restraints and (4) positional restraints. The equilibrium target values and force constants can be specified as either a float or Pint quantity through the openff-units wrapper.
(1) Static Restraints
Static restraints do not change during the whole APR process and do not affect the free energy. We apply static restraints
on the host (or protein) molecule to orient the host/protein degrees of freedom. The static restraints are composed of
distance, angle, and torsional (DAT) restraints based on the choice of anchor atoms. For host-guest systems, we need to
define three anchor atoms [H1,H2,H3] and combined with three dummy atoms [D1,D2,D3], we apply a total of six
static restraints on the host molecule (three for the translation and three for orientation).
To generate static restraints we use the function static_DAT_restraints. As an example, to apply a distance restraint
on D1 and H1 with a force constant of 5 kcal/mol/\(Å^2\) we call
from openff.units import unit
from paprika.restraints import static_DAT_restraint
k_dist = 5.0 * unit.kcal / unit.mole / unit.angstrom ** 2
dist_static = static_DAT_restraint(
restraint_mask_list = [D1, H1],
num_window_list = windows, # list: [len(attach_lambda), len(pull_windows), len(release_lambda)]
ref_structure = structure, # Structure file (PDB) or ParmEd structure object
force_constant = k_dist,
)
The equilibrium target for the harmonic restraint is estimated from the ref_structure.
(2) Varying Restraints
As the name suggests, these restraints change during the APR process. During the attach and release phases, the force constants of these restraints changes. In the pull phase, varying restraints can have their equilibrium position change, and this can be used as the restraint to pull the guest molecule out of the host molecule.
To generate varying restraints, we use the DAT_restraint class. The code below shows a restraints r that starts
from 6.0 Å to 24 Å in the pull phase and stays restrained at 24 Å during the release phase.
from paprika.restraints import DAT_restraint
r_init = 6.0 * unit.angstrom
r_final = 24.0 * unit.angstrom
k_dist = 5.0 * unit.kcal / unit.mole / unit.angstrom ** 2
r = DAT_restraint()
r.mask1 = D1
r.mask2 = G1
r.topology = structure
r.auto_apr = True
r.continuous_apr = True
r.attach["target"] = r_init
r.attach["fraction_list"] = attach_lambda
r.attach["fc_final"] = k_dist
r.pull["target_final"] = r_final
r.pull["num_windows"] = len(pull_windows)
r.release["target"] = r_final
r.release["fraction_list"] = [1.0] * len(release_lambda)
r.release["fc_final"] = k_dist
r.initialize()
Note
The DAT_restraint class can also be used to apply conformational restraints on the host and/or guest molecule.
For example, distance “jack” and dihedral restraints can be applied to cucurbiturils and cyclodextrins host molecules,
respectively, to make the binding site more accessible.
(3) Wall Restraints (optional)
Wall restraints are half-harmonic potentials that is useful for preventing guest molecules from leaving the binding
site (for weak binding) or preventing the guest molecule from flipping during the attach phase. We still use the
DAT_restraint class to generate the restraints but will use the custom_restraint_values method to generate
the half-harmonic potential.
Note
custom_restraint_values follows the AMBER NMR-restraint format, see Chapter 27 in the AMBER20 manual
for more details.
Below is an example for generating a “lower wall” restraint that prevents the angle of [D2,G1,G2] from
decreasing below 91 degrees.
r_wall = 91.0 * unit.degrees
k_wall = 200.0 * unit.kcal / unit.mole / unit.radians ** 2
wall_orient = DAT_restraint()
wall_orient.mask1 = D1
wall_orient.mask2 = G1
wall_orient.mask3 = G2
wall_orient.topology = structure
wall_orient.auto_apr = True
wall_orient.continuous_apr = True
wall_orient.attach["num_windows"] = attach_fractions
wall_orient.attach["fc_initial"] = k_wall
wall_orient.attach["fc_final"] = k_wall
wall_orient.custom_restraint_values["r1"] = k_wall
wall_orient.custom_restraint_values["r2"] = 0.0
wall_orient.custom_restraint_values["rk2"] = k_wall
wall_orient.custom_restraint_values["rk3"] = 0.0
wall_orient.initialize()
(4) Positional Restraints
Positional restraints in APR simulations are applied to the dummy atoms. Together with static restraints, this
provides a laboratory frame of reference for the host-guest complex. Different MD programs handles positional restraints
differently. For example, in AMBER you can define positional restraints in the input configuration file using the
ntr keyword (Chapter 19 in the AMBER20 manual). For other programs like GROMACS and NAMD that uses Plumed,
positional restraints can be applied using the method add_dummy_atom_restraints().
Note
tleap may shift the coordinates of the system when it solvates the structure. Applying the positional restraints
before the solvating the structure may lead to undesired errors during simulations. Therefore, special care needs to
be taken when applying positional restraints. Take a look at tutorials 5
and 6 to see this distinction.
Creating the APR windows and saving restraints to file
To create the windows for the APR calculation we need to parse a varying restraint to the utility function create_window_list.
This function will return a list of strings for the APR protocol
window_list = create_window_list(restraints_list)
window_list
["a000", "a001", ..., "p000", "p001", ...]
It may also be useful to save both the windows list and the restraints to a JSON file so you do not need to redefine again.
The restraints can be saved to a JSON file using the utility function save_restraints.
from paprika.io import save_restraints
save_restraints(restraints_list, filepath="restraints.json")
import json
with open("windows.json", "w") as f:
dumped = json.dumps(window_list)
f.write(dumped)
Extending/adding more windows
Sometimes it may be necessary to add more windows in the APR calculation due to insufficient overlap between neighboring windows. For convenience we can add the windows at the end of the current list instead of inserting them in order. For example, let’s say that we have a defined a restraint that spans from 8.4 to 9.8 Å and we want to add three windows between 8.6 and 9.0 Å.
r_restraint.pull
{'fc': 10.0,
'target_initial': None,
'target_final': None,
'num_windows': None,
'target_increment': None,
'fraction_increment': None,
'fraction_list': None,
'target_list': array([8.4, 8.6, 9. , 9.4, 9.8])}
We will just need to append the target_list of this dictionary and reinitialize the restraints
r_restraint.pull["target_list"] = np.append(r_restraint.pull["target_list"], [8.7, 8.8, 8.9])
r_restraint.initialize()
r_restraint.pull
{'fc': 10.0,
'target_initial': None,
'target_final': None,
'num_windows': None,
'target_increment': None,
'fraction_increment': None,
'fraction_list': None,
'target_list': array([8.4, 8.6, 9. , 9.4, 9.8, 8.7, 8.8, 8.9])}
We can save the updated restraints to a new file and pass it to the analysis script. The fe_calc class will take
care of the window ordering thus there is no need to manually order the windows.
Running a Simulation¶
paprika provides wrappers with the Simulate module for a number of MD engines enabling us to run the simulations
in python.
from paprika.simulate import AMBER
simulation = AMBER()
simulation.executable = "pmemd.cuda"
simulation.gpu_devices = "0"
simulation.path = "simulation"
simulation.prefix = "equilibration"
simulation.coordinates = "minimize.rst7"
simulation.ref = "host-guest-dum.rst7"
simulation.topology = "host-guest-dum.prmtop"
simulation.restraint_file = "disang.rest"
simulation.config_pbc_md()
# Positional restraints on dummy atoms
simulation.cntrl["ntr"] = 1
simulation.cntrl["restraint_wt"] = 50.0
simulation.cntrl["restraintmask"] = "'@DUM'"
print(f"Running equilibration in window {window}...")
simulation.run()
Analysis¶
Once the simulation is complete, the free energy can be obtained using the Analysis module, which will also
estimate the uncertainties using the bootstrapping method. There are three types of methods that you can do
with the Analysis module: (1) thermodynamic integration with block-data analysis (“ti-block”), (2) multistate
Benett-Acceptance-Ratio with block-data analysis (“mbar-block”), and (3) multistate Benett-Acceptance-Ratio with
autocorrelation analysis (“mbar-autoc”).
from paprika.analysis import fe_calc
from paprika.io import load_restraints
restraints_list = load_restraints(filepath="restraints.json")
free_energy = fe_calc()
free_energy.prmtop = "host-guest-dum.prmtop"
free_energy.trajectory = 'production.nc'
free_energy.path = "windows"
free_energy.restraint_list = restraints_list
free_energy.collect_data()
free_energy.methods = ['ti-block']
free_energy.ti_matrix = "full"
free_energy.bootcycles = 1000
free_energy.compute_free_energy()
We can also estimate the free energy cost of releasing the restraints on the guest molecule semianalytically. To do
this we need to extract the restraints that is specific to the guest molecule. The extract_guest_restraints
function from the restraints module and pass this to the analysis object.
import parmed as pmd
from paprika.restraints.utils import extract_guest_restraints
structure = pmd.load_file("guest.prmtop", "guest.rst7", structure=True)
guest_restraints = extract_guest_restraints(structure, restraints_list, guest_resname="GST")
free_energy.compute_ref_state_work(guest_restraints)
The results are stored in the variable results as a python dictionary and you can save this to a JSON file.
print(free_energy.results["pull"]["ti-block"]["fe"])
-3.82139135698 kcal/mol
free_energy.save_results("APR_results.json")
The processed simulation data can also be saved to a JSON file so that you do not need to re-read the MD trajectories if you need to do further analysis.
free_energy.save_data("APR_simulation_data.json")
free_energy.collect_data_from_json("APR_simulation_data.json")