
pAPRika is a python toolkit for setting up, running, and analyzing free energy molecular dynamics simulations.
Theory¶
Binding free energy¶
In the attach-pull-release (APR) method, a guest molecule is physically pulled out of the host molecule along a defined path by applying harmonic restraints. The binding free energy is then computed in terms of the sum of the work required for (1) attaching restraints to the bound complex, (2) pulling the guest molecule out of the host molecule and (3) releasing the restraints of the unbound complex:
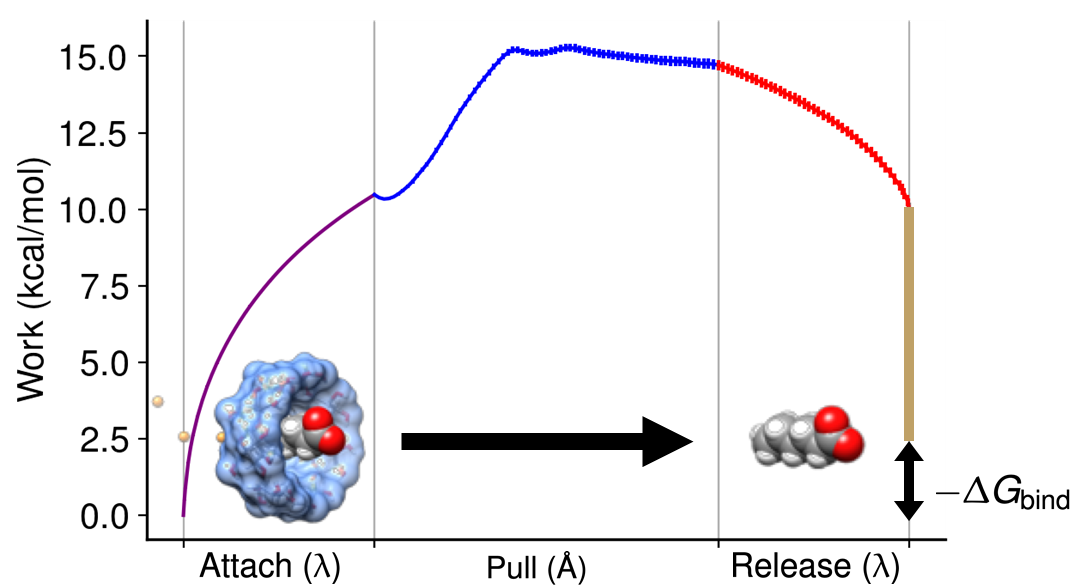
In the equation above, the work of releasing the restraints is split into two: \(W_\text{release-conf}\) and \(W_\text{release-std}\). These represents the work for releasing conformational restraints (applied to host or guest molecules) and the guest’s translational and rotational restraints to the standard concentration, respectively. The translational and rotational degrees of freedom of the guest molecule is defined in spherical coordinates \((r,\theta,\phi)\) and Euler angles \((\alpha,\beta,\gamma)\), respectively. Unlike the work for releasing conformational restraints, the work for releasing the guest’s restraints to the standard concentration can be evaluated analytically:
For other terms in equation (1), the calculation is broken up into a series of independent windows. During the attach and release phase, the force constant of the restraints increases and decreases, respectively. In the pull phase, the distance between the guest and host is discretized from point A (bound) to point B (unbound) by changing the equilibrium position of the distance restraint. The work for each phase can be estimated using either thermodynamic integration (TI) or the multistate-Bennett-Acceptance-Ratio (MBAR).
pAPRika estimates the standard error of the mean (SEM) with either block data analysis or autocorrelation.
Binding enthalpy¶
The binding enthalpy is the difference in the partial molar enthalpies of the bound complex and the separated molecules. Setting up binding enthalpy calculations is more straightforward than the binding free energy if we use the direct method. In the direct method, the binding enthalpy is obtained from the difference in the mean potential energy of the bound and unbound complex.
In the context of APR simulations, the mean potential energy is estimated from the first and last window. These windows, however, will need to be run for much longer until the fluctuation of the mean potential energy decreases below a desired value. Another way to estimate the binding enthalpy is with the van’t Hoff method:
Here, the binding enthalpy is obtained by a linear regression of \(\text{ln}\,K_\text{eq}\) vs \(1/T\). The equilibrium constant will need to be evaluated at different temperatures in order the estimate the binding enthalpy with the van’t Hoff method.
Supported Molecular Dynamics Engines¶
Currently, we can use pAPRika to set up, perform, and analyze APR simulations with the AMBER, OpenMM, GROMACS, and NAMD programs. pAPRika supports these MD programs by providing a python-based wrapper for running the simulations except for OpenMM. These MD programs provide their own interface for restraints (e.g., AMBER uses NMR-style restraints). However, pAPRika also provides modules for generating APR restraints in Plumed and Colvars format, which can interface with various MD programs.
Future releases of pAPRika will include support for other MD engines like LAMMPS. Plumed is supported by all of the listed MD programs below, but for NAMD, only NVT simulations can be performed. For running NPT simulations in NAMD, however, we can use the Colvars module. Also, a pipeline for running double-decoupling calculations with pAPRika is currently in development and will be released soon.
MD Engine
|
Restraints Module
|
|||
|---|---|---|---|---|
NMR
|
XML
|
Plumed
|
Colvars
|
|
AMBER
|
✓
|
✕
|
✓
|
✕
|
OpenMM
|
✕
|
✓
|
✓**
|
✕
|
GROMACS
|
✕
|
✕
|
✓
|
✕
|
NAMD
|
✕
|
✕
|
✓
|
✓
|
LAMMPS*
|
✕
|
✕
|
✓*
|
✓*
|
* Currently not supported but will be available in future releases.
** Supported but have not yet been tested.
License¶
pAPRika is licensed under the BSD 3-Clause license.
References¶
Velez-Vega, C. & Gilson, M. K. Overcoming Dissipation in the Calculation of Standard Binding Free Energies by Ligand Extraction. J. Comput. Chem. 34, 2360–2371 (2013).
Fenley, A. T., Henriksen, N. M., Muddana, H. S. & Gilson, M. K. Bridging calorimetry and simulation through precise calculations of cucurbituril-guest binding enthalpies. J. Chem. Theory Comput. 10, 4069–4078 (2014).
Henriksen, N. M., Fenley, A. T. & Gilson, M. K. Computational Calorimetry: High-Precision Calculation of Host−Guest Binding Thermodynamics. J. Chem. Theory Comput. 11, 4377–4394 (2015).